

Die Medizinproduktebranche ist streng reguliert und unterliegt hohen Anforderungen an Produkthaftung und Sicherheit. Für Hersteller und Anbieter bedeutet dies komplexe gesetzliche Vorgaben, die eine sorgfältige Einhaltung erfordern. Wir möchten Ihnen hier einen Überblick über die wesentlichen Regulierungen und Anforderungen geben – von der Entwicklung und Zulassung bis hin zur Überwachung und Haftung im Markt.
Regulatorische Anforderungen und Haftung bei Medizinprodukten
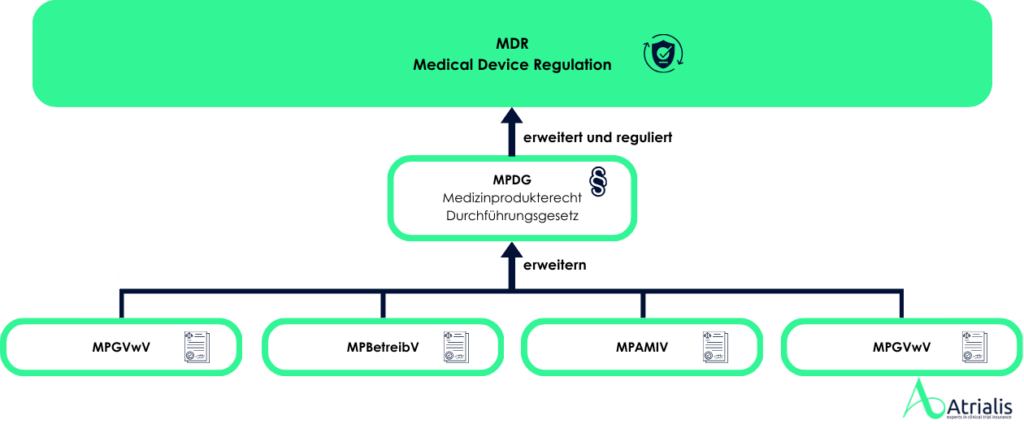
Die europäische Medizinprodukteverordnung MDR (Medical Device Regulation, EU 2017/745) überlässt es den Mitgliedsstaaten spezifische Haftungsbestimmungen einzuführen. In Deutschland regelt das Medizinproduktedurchführungsgesetz (MPDG) diese Thematik, schreibt jedoch eine Produkthaftpflichtversicherung nicht vor. Wir empfehlen Ihnen jedoch dringend, diese Absicherung freiwillig vorzunehmen. Ein umfassender Versicherungsschutz ist ein entscheidender Faktor für die Sicherheit und Zukunft Ihres Unternehmens.
Für Unternehmen in der Medizinproduktebranche ist es entscheidend, einen Versicherungsschutz zu haben, der ihre spezifischen Risiken abdeckt. Besonders im Rahmen der MDR übernehmen Händler und Importeure unter bestimmten Bedingungen die Haftungspflichten der Hersteller – beispielsweise, wenn sie Produkte unter ihrem eigenen Markennamen auf den Markt vertreiben. Diese erweiterten Haftungspflichten machen deutlich, wie wichtig ein umfassender Versicherungsschutz für alle Wirtschaftsakteure in der Lieferkette ist.
Warum wird eine spezielle Versicherung für Medizinprodukte benötigt?
Obwohl das MPDG in Deutschland keine Pflicht zur Produkthaftpflichtversicherung vorschreibt, schließen viele Unternehmen freiwillig eine solche ab, um sich gegen Haftungsansprüche abzusichern. Medizinprodukte, von einfachen Verbrauchsmaterialien wie Kanülen bis hin zu komplexen implantierbaren Herzschrittmachern, erfordern aufgrund ihrer unterschiedlichen Risikoklassen eine risikogerechte Einordnung, um den passenden Versicherungsschutz zu gewährleisten. Ein maßgeschneiderter Versicherungsschutz kann für Unternehmen in der Medizinproduktebranche existenzsichernd sein, da die möglichen Haftungsrisiken enorme finanzielle Auswirkungen haben können.
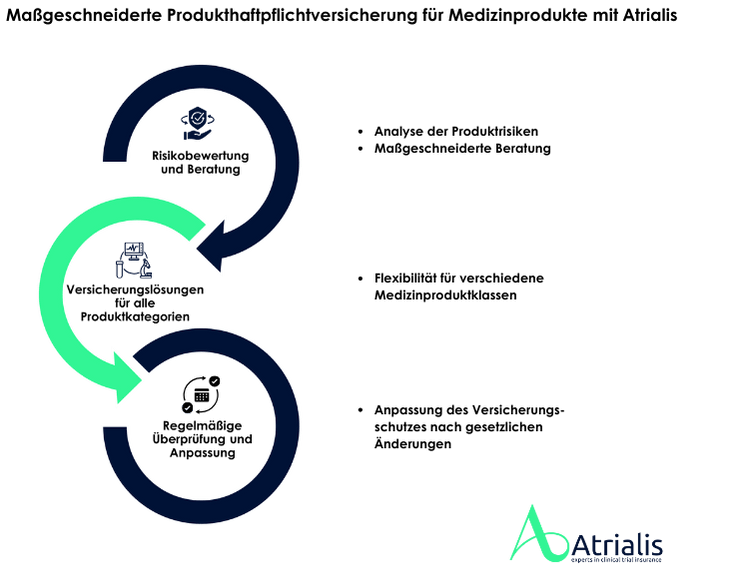
Unsere Lösung: Maßgeschneiderte Produkthaftpflichtversicherung für Medizinprodukte
Wir bieten eine Medizinproduktehaftpflichtversicherung an, die gezielt auf die spezifischen Anforderungen und Risiken Ihrer Produkte abgestimmt ist. Unsere Leistungen umfassen:
Ob einfache Verbrauchsmaterialien oder Hightech-Implantate – wir sorgen für den passenden Deckungsschutz und unterstützen Sie dabei, die regulatorischen Anforderungen der MDR und des MPDGs zu erfüllen.
Lassen Sie uns gemeinsam sicherstellen, dass Ihre Produkte und Ihr Unternehmen optimal abgesichert sind.

"Bei der Produkthaftpflichtversicherung für Medizinprodukte zählt nicht nur die Wahl der passenden Versicherungssumme – die Qualität der Bedingungen ist entscheidend. Nur klare und umfassende Regelungen garantieren im Ernstfall optimalen Schutz. Mit unseren speziell entwickelten Versicherungsbedingungen bieten wir unseren Mandanten marktführenden Schutz."
Florian Eckstein - Geschäftsführender Gesellschafter Atrialis GmbH



3. Was kostet eine Produkthaftpflichtversicherung?
Die Kosten für eine Produkthaftpflichtversicherung können stark variieren und hängen von mehreren Faktoren ab, insbesondere vom Risiko des Produkts, der Umsatzgröße und Wahl der Versicherungssumme. Bei geringen Risiken und vor Kommerzialisierung im breiten Markt können die Prämien für kleinere Unternehmen ab 850 bis 1.100 EUR pro Jahr beginnen.
Für Unternehmen, die Medizinprodukte herstellen oder bereits kommerziell vertreiben, steigen die Kosten abhängig vom Umsatz schnell auf 2.500 EUR jährlich oder sogar mehr. Zu den entscheidenden Faktoren, die die Prämie beeinflussen, gehören:
Um eine präzise Einschätzung Ihres Absicherungsbedarfs und realistischen Prämienschätzungen zu erhalten, sollten Sie Ihre Produkte und spezifischen Risikofaktoren durch einen spezialisierten Versicherungsmakler oder Anbieter bewerten lassen. Sprechen Sie uns hierzu gerne an!

Laut einer Umfrage des DIHK und SPECTARIS sehen fast 80% der Medizintechnikunternehmen aufgrund der EU-Verordnung für Medizinprodukte (MDR) erhebliche Schwierigkeiten bei der Markteinführung von Produkten.

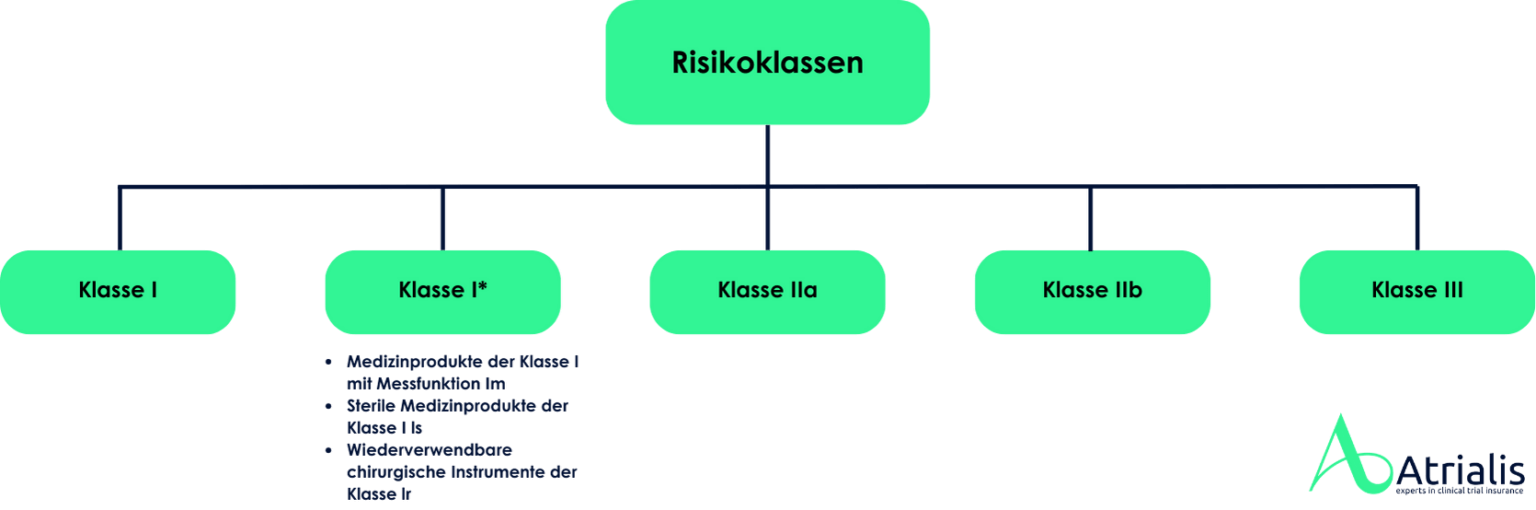
In Europa werden Medizinprodukte in der Regel eigenverantwortlich durch den Hersteller auf den Markt gebracht. Dabei ist der Hersteller verpflichtet, ein Konformitätsbewertungsverfahren für die Produkte durchzuführen. Zur Durchführung der Konformitätsbewertung ist es gemäß MDR erforderlich, Medizinprodukte einer entsprechenden Risikoklasse zuzuordnen. Diese Klassifizierung erfolgt auf Grundlage der Zweckbestimmung des Produkts und der damit verbundenen potenziellen Risiken für Patienten, Anwender und Dritte.
Die MDR unterscheidet zwischen vier Risikoklassen (Klasse I, IIa, IIb und III). Je nach Risikoklasse variieren die Anforderungen an die Konformitätsbewertung, wobei höhere Risikoklassen strengere Prüf- und Nachweispflichten mit sich bringen. Für viele Produkte ist die Einbeziehung einer Benannten Stelle obligatorisch, die die Einhaltung der Vorschriften prüft und gegebenenfalls ein Konformitätszertifikat ausstellt. Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) führt kein Verzeichnis über die Klassifizierung von in Europa in Verkehr gebrachten Medizinprodukten. Pauschale Entscheidungen zu gesamten Produktgruppen sind ebenfalls nicht möglich, da die Risikoklassifizierung stets auf der individuellen Zweckbestimmung des Produkts sowie den spezifischen Angaben des Herstellers basiert.
Ein wesentlicher Bestandteil der Konformitätsbewertung ist die Erstellung der technischen Dokumentation, welche detaillierte Informationen über das Produkt, seine Herstellung, die Risikomanagementprozesse und die klinische Bewertung umfasst. Darüber hinaus müssen Hersteller sicherstellen, dass ihr Qualitätsmanagementsystem den Anforderungen der MDR entspricht, bspw. durch eine ISO 13485-Zertifizierung.
Ein weiteres zentrales Element für die Konformitätsbewertung ist die CE-Kennzeichnung. Diese wird auf dem Produkt oder auf seiner (sterilen) Verpackung angebracht und zeigt an, dass das Produkt die grundlegenden Sicherheits- und Leistungsanforderungen der MDR erfüllt und auf dem europäischen Markt in Verkehr gebracht werden darf. Sie muss auch in der Gebrauchsanweisung und auf der Handelsverpackung erscheinen. Dabei gibt es genaue Vorgaben bezüglich der Größe und Proportion der CE-Kennzeichnung, die in den entsprechenden Normen festgelegt sind.
➔ Sind diese Vorgaben erfüllt, gilt das Medizinprodukt als „MDR-konform„.
Die Entwicklung und der Einsatz von Medizintechnik bringen immense Fortschritte für die Gesundheitsversorgung, bergen aber auch Risiken – sei es durch technische Defekte, Anwendungsfehler oder regulatorische Anforderungen. Unternehmen in der Medizintechnikbranche stehen daher vor der Herausforderung, sich gegen vielfältige Risiken abzusichern. Da der Versicherungsschutz je nach Produktart, Marktregion und regulatorischen Anforderungen stark variieren kann, sind maßgeschneiderte Lösungen entscheidend. Wir beraten Sie gerne zu individuellen Versicherungslösungen für Ihr Medizinprodukt!
Wir haben ein spezielles Versicherungskonzept für digitale Medizinprodukte entwickelt, wie etwa SaMD (Software as Medical Device) – Produkte, bei denen wir im Markt derzeit einen Megatrend beobachten.
In unserer innovativen und marktführenden Versicherungslösung „Digital Health Protect by Atrialis“ ist nicht nur die obligatorische Produkthaftung in Anlehnung an die geltenden gesetzlichen und regulatorischen Anforderungen nach MDR abgesichert, sondern auch eine Kombination mit einer IT-Vermögensschadenhaftpflicht (Tech PI), die bei allgemeinen Ansprüchen finanzieller Schäden aus bspw. Fehlern oder Bugs der Software resultieren.
Software as a Medical Device (SaMD) bezeichnet Software, die eigenständig – also ohne ein physisches medizinisches Gerät – medizinische Zwecke erfüllt. Dazu gehören z. B. Programme zur Diagnose, Therapieplanung oder Gesundheitsüberwachung. Je nach Markt müssen SaMD-Produkte unterschiedliche regulatorische Anforderungen erfüllen. In Europa unterliegen SaMD der Medical Device Regulation (MDR). Typische Anwendungsbereiche sind Algorithmen zur Krebserkennung (z.B. durch Bildanalyse), Apps zur Überwachung von chronischen Erkrankungen, AI-gestützte Diagnosetools oder Digitale Therapeutika (DTx).
Besonders AI-powered oder AI-assisted SaMD sind mit in unserem “Digital Health Protect by Atrialis“ besonders geschützt, da aktuellen Standard-Versicherungsbedingungen am Versicherungsmarkt bisher keine Klarstellungen oder sogar Lücken für moderne und innovative Software-as-a-Medical Device-Produkte und den Einsatz von KI aufweisen. Fragen Sie nach unserer eigenen “Digital Health Protect by Atrialis“-Lösung für Ihre CE-Zulassung oder Vertriebschancen in Europa, um haftungssicher und compliant nach MDR zu sein.
Als spezialisierter Versicherungsmakler für medizinische Softwarelösungen kennen wir die Risiken, regulatorischen Herausforderungen und Haftungspotenziale von SaMD. Wir bieten Ihnen passgenaue Absicherung – national und international.
Die medizinische Zweckbestimmung eines Produkts beschreibt den medizinischen Zweck, für den ein Produkt entwickelt und bestimmt ist. Es geht darum, welche medizinische Funktion das Produkt erfüllen soll. Dies umfasst unter anderem:
Die Zweckbestimmung beschreibt also, in welchen Bereichen (wie z. B. der Diagnostik oder Therapie) und mit welchen Zielen (z. B. Heilung, Unterstützung oder Prävention) das Produkt eingesetzt wird.
Der Wirkmechanismus eines Medizinprodukts beschreibt die Art und Weise, wie das Produkt funktioniert. In der MDR wird dabei speziell darauf hingewiesen, dass der Wirkmechanismus nicht auf pharmakologischen, immunologischen oder metabolischen Effekten beruht. Das bedeutet, das Produkt wirkt nicht über die Einnahme von Medikamenten oder über die Beeinflussung von Körperfunktionen wie das Immunsystem oder den Stoffwechsel. Stattdessen funktioniert es über physikalische oder chemische Prozesse.
Ein Beispiel: ein Herzschrittmacher nutzt physikalische Effekte, um das Herz zu stimulieren, aber er beeinflusst nicht direkt den Stoffwechsel oder das Immunsystem.
Zubehör sind Produkte, die zwar selbst keine Medizinprodukte sind, aber vom Hersteller speziell dafür entwickelt wurden, zusammen mit einem oder mehreren Medizinprodukten verwendet zu werden. Das Zubehör unterstützt die Nutzung des Hauptprodukts und hilft, die medizinische Funktion des Produkts zu optimieren. Es ist also kein eigenständiges medizinisches Produkt, sondern eher eine Ergänzung, um das Hauptprodukt funktionsfähig zu machen oder dessen Effizienz zu steigern.
Das Zubehör kann dazu dienen, die Zweckbestimmung des Medizinprodukts zu gewährleisten, es für spezielle Anwendungen anzupassen, die Handhabung zu erleichtern, die Leistungsfähigkeit zu steigern oder die Funktionen des Produkts mit denen anderer Geräte zu kombinieren.
Ein Schlauch zum Beispiel, der in ein medizinisches Gerät wie ein Beatmungsgerät integriert wird, ist Zubehör. Ohne den Schlauch würde das Gerät nicht wie vorgesehen funktionieren.
„Aktive Produkte“ sind Medizinprodukte, deren Betrieb eine externe Energiequelle benötigt. Diese Energiequelle kann nicht die menschliche Körperkraft oder Schwerkraft sein. Diese Produkte erzeugen eine aktive Funktion, die für den medizinischen Zweck des Produkts notwendig ist. Ein Beispiel:
„Systeme und Behandlungseinheiten“ bestehen aus einer Kombination von Medizinprodukten, die zusammen verwendet werden, um einen bestimmten medizinischen Zweck zu erfüllen. Diese Produkte arbeiten zusammen, um eine komplexe medizinische Funktion zu unterstützen.
Ein Beispiel: Ein Operationssystem könnte aus mehreren Instrumenten bestehen, wie z. B. einem chirurgischen Roboter, Kameras und anderen unterstützenden Geräten, die zusammenarbeiten, um eine Operation durchzuführen.
Solche Systeme und Behandlungseinheiten können mehrere Produkte umfassen, die in einer bestimmten Art und Weise miteinander verbunden oder integriert sind, um den Behandlungserfolg zu sichern.
Die „Zweckbestimmung“ eines Produkts bezieht sich auf die Nutzung, die der Hersteller für das Produkt vorgesehen hat. Diese wird auf der Kennzeichnung des Produkts und in der Gebrauchsanweisung sowie in Werbe- oder Verkaufsangaben definiert. Sie beschreibt, was das Produkt tun soll und wie es verwendet wird, wie zum Beispiel:
Die Zweckbestimmung gibt also an, in welchem medizinischen Kontext das Produkt verwendet werden soll und welches Ziel der Einsatz hat.
Im Rahmen der MDR gibt es festgelegte Verfahren, um zu klären, wie ein Produkt klassifiziert und behandelt werden soll. Die Feststellung des Status eines Produkts erfolgt durch die Europäische Kommission. Sie entscheidet, ob ein Produkt ein Medizinprodukt ist und welchen Status es hat.
Die Statusfeststellung kann auf folgende Weise initiiert werden:
• Auf Anfrage eines Mitgliedstaates: Ein Mitgliedstaat kann bei der Kommission eine Überprüfung anfordern, wenn es einen guten Grund dafür gibt (z. B. Unklarheiten über die Klassifizierung des Produkts).
• Aus eigener Initiative der Kommission:Die Kommission kann auch selbstständig eine Überprüfung anstoßen, wenn sie der Meinung ist, dass dies nötig ist (z. B. bei neuen Produkten oder Technologien).
• Nach Anhörung der Koordinierungsgruppe Medizinprodukte: Bevor die Kommission eine endgültige Entscheidung trifft, wird sie eine Expertengruppe (Koordinierungsgruppe) konsultieren, die sie berät.
Diese Schritte dienen dazu, Rechtssicherheit zu gewährleisten und sicherzustellen, dass Produkte korrekt klassifiziert werden, was wiederum die Sicherheit und den Schutz der Gesundheit der EU-Bürger gewährleistet.