

The medical device industry is strictly regulated and subject to high product liability and safety requirements. For manufacturers and suppliers, this means complex legal requirements that require careful compliance. We would like to give you an overview of the key regulations and requirements - from development and approval to monitoring and liability in the market.
Regulatory requirements and liability for medical devices
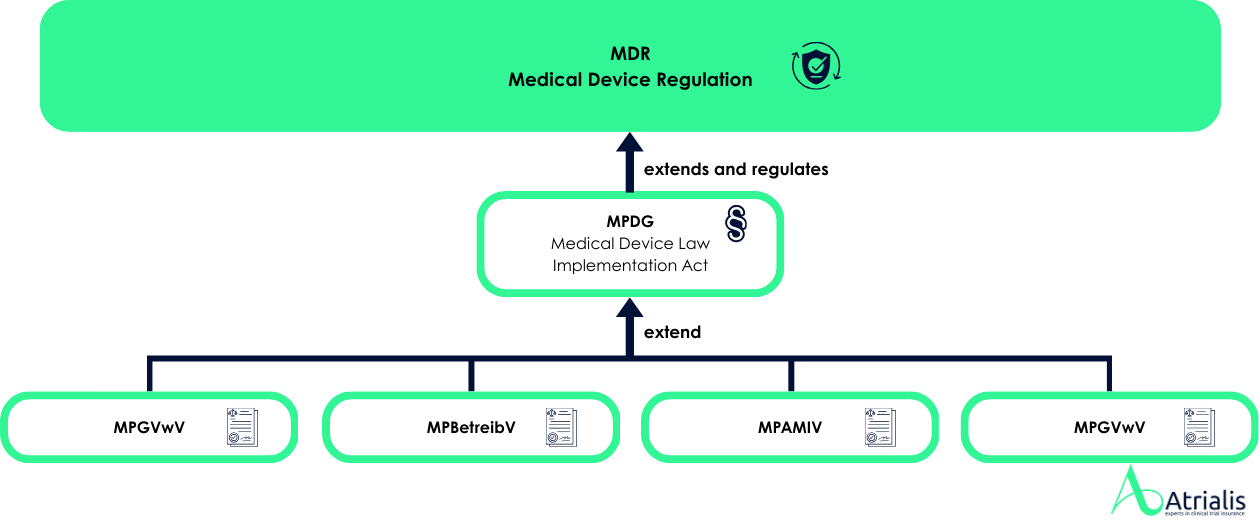
The European Medical Device Regulation ( MDR , EU 2017/745)leaves it up to the member states to introduce specific liability provisions. In Germany, the Medical Devices Implementation Act (MPDG) regulates this topic, but does not require product liability insurance. However, we strongly recommend that youtake out this cover voluntarily. Comprehensive insurance cover is a decisive factor for the safety and future of your company.
It is crucial for companies in the medical device industry to have insurance cover that covers their specific risks covered. Under the MDR in particular, distributors and importers assume the liability obligations of manufacturers under certain conditions - for example, if they sell products on the market under their own brand name. These extended liability obligations highlight the importance of comprehensive insurance cover for all economic operators in the supply chain.
Why is special insurance needed for medical devices?
Although the MPDG in Germany no obligation for product liability insurance product liability insurance in Germany, many companies voluntarily to protect themselves against liability claims. Medical devices, from simple consumables such as cannulas to complex implantable pacemakers, require a risk-based classification due to their different risk classes in order to ensure suitable insurance cover. Tailor-made insurance cover can secure the existence of companies in the medical device industry, as the potential liability risks can have enormous financial consequences.

Our solution: Tailor-made product liability insurance for medical devices
We offer medical device liability insurance that is tailored to the specific requirements and risks of your products. Our services include:
Whether simple consumables or high-tech implants - we provide the right cover and support you in meeting the regulatory requirements of the MDR and the MPDG.
Let us work together to ensure that your products and your company are optimally covered.
We let our work and our clients speak for themselves.

“In response to a short-notice request for proposals for product liability insurance, Atrialis GmbH met our requirements quickly and precisely. Their advice was consistently knowledgeable, transparent, and solution-oriented. We were particularly impressed by the speed and quality of the market feedback. We found the collaboration to be very professional and efficient.”



3. what does product liability insurance cost?
The cost of product liability insurance can vary greatly and depends on several factors, in particular the risk of the product, the size of turnover and the choice of sum insured. For low risks and before commercialization in the broad market, premiums for smaller companies can start from EUR 850 to EUR 1,100 per year.
For companies that manufacture medical devices or already distribute them commercially, the costs quickly rise to EUR 2,500 per year or even more, depending on turnover. The decisive factors that influence the premium include
In order to obtain a precise assessment of your insurance needs and realistic premium estimates, you should have your products and specific risk factors evaluated by a specialized insurance broker or provider. Please do not hesitate to contact us!

According to a survey conducted by the DIHK and SPECTARIS, almost 80% of medical technology companies see considerable difficulties in launching products on the market due to the EU Medical Device Regulation (MDR).

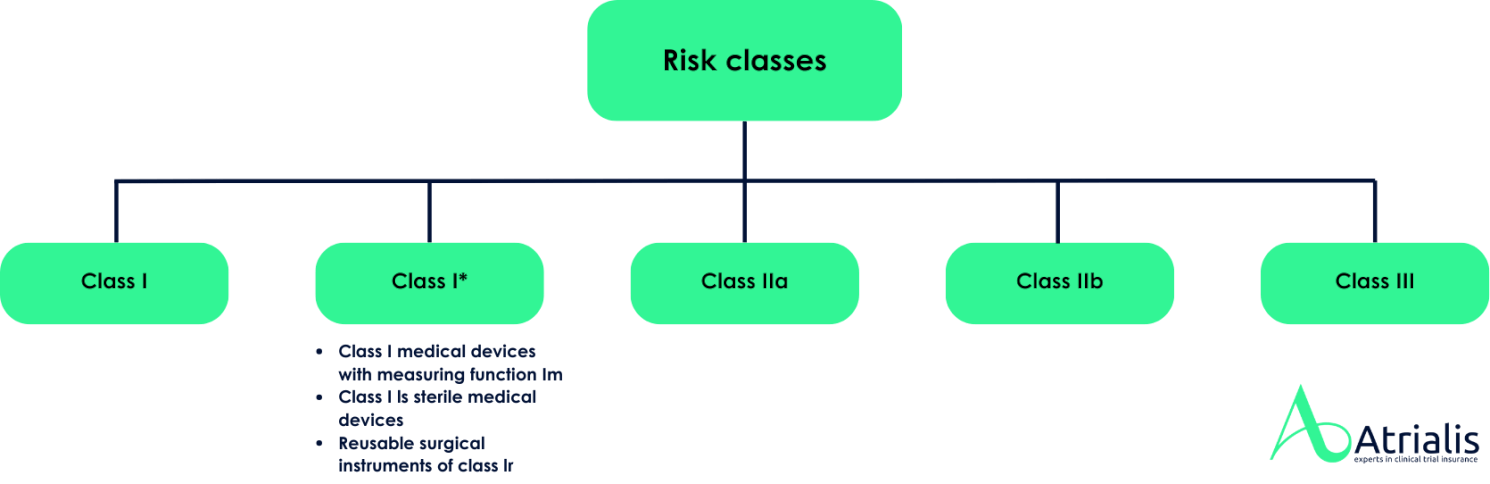
In Europe, medical devices are generally placed on the market under the manufacturer's own responsibility. The manufacturer is obliged to carry out a conformity assessment procedure for the products. To carry out the conformity assessment, medical devices must be assigned to a corresponding risk class in accordance with the MDR . This classification is based on the intended purpose of the device and the associated potential risks for patients, users and third parties.
The MDR distinguishes between four risk classes (Class I, IIa, IIb and III). The conformity assessment requirements vary depending on the risk class, with higher risk classes entailing stricter testing and verification obligations. For many products, the involvement of a notified body is mandatory, which checks compliance with the regulations and issues a certificate of conformity if necessary. The Federal Institute for Drugs and Medical Devices (BfArM) does not maintain no register on the classification of medical devices placed on the market in Europe. Blanket decisions on entire product groups are also not possible, as the risk classification is always based on the individual intended purpose of the product and the specific information provided by the manufacturer.
An essential part of the conformity assessment is the preparation of the technical documentation, which includes detailed information about the product, its manufacture, the risk management processes and the clinical evaluation. In addition, manufacturers must ensure that their quality management system meets the requirements of the MDR, e.g. through ISO 13485 certification.
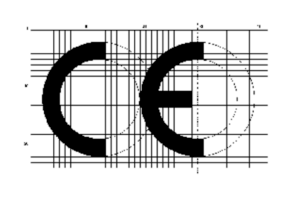
Another key element for the conformity assessment is the CE certificate. This is affixed to the product or its (sterile) packaging and indicates that the product meets the essential safety and performance requirements of the MDR and may be placed on the European market. It must also appear in the instructions for use and on the retail packaging. There are precise specifications regarding the size and proportions of the CE marking, which are set out in the relevant standards.
➔ If these requirements are met, the medical device is considered "MDR-compliant".
The development and use of medical technology brings immense progress for healthcare, but also entails risks - whether due to technical defects, application errors or regulatory requirements. Companies in the medical technology sector are therefore faced with the challenge of insuring themselves against a wide range of risks. As insurance cover can vary greatly depending on the type of product, market region and regulatory requirements, tailor-made solutions are crucial. We will be happy to advise you on individual insurance solutions for your medical device!
We have developed a special insurance concept for digital medical devices, such as SaMD (Software as Medical Device) - products for which we are currently observing a megatrend in the market.
Our innovative and market-leading insurance solution "Digital Health Protect by Atrialis" not only covers mandatory product liability in accordance with the applicable legal and regulatory requirements under the MDR, but also a combination with IT property damage liability (Tech PI), which covers general claims for financial losses resulting from, for example, errors or bugs in the software.
Software as a Medical Device (SaMD) refers to software that fulfills medical purposes independently - i.e. without a physical medical device. This includes, for example, programs for diagnosis, therapy planning or health monitoring. Depending on the market, SaMD products must fulfill different regulatory requirements. In Europe, SaMDs are subject to the Medical Device Regulation(MDR). Typical areas of application are algorithms for cancer detection (e.g. through image analysis), apps for monitoring chronic diseases, AI-supported diagnostic tools or digital therapeutics (DTx).
AI-powered or AI-assisted SaMD in particular are included in our "Digital Health Protect by Atrialis" as current standard insurance terms and conditions on the insurance market do not yet have any clarifications or even gaps for modern and innovative software-as-a-medical device products and the use of AI. Ask for our own "Digital Health Protect by Atrialis" solution for your CE approval or distribution opportunities in Europe to be liability-proof and compliant according to MDR.
As a specialized insurance broker for medical software solutions, we know the risks, regulatory challenges and liability potential of SaMD. We offer you tailor-made cover - nationally and internationally.

"When it comes to product liability insurance for medical devices, it's not just the choice of the right sum insured that counts - the quality of the terms and conditions is crucial. Only clear and comprehensive regulations guarantee optimum protection in an emergency. With our specially developed insurance conditions, we offer our clients market-leading protection."
Florian Eckstein - Managing Partner Atrialis GmbH
The medical purpose of a product describes the medical purpose for which a product is developed and intended. It is about the medical function the product is intended to fulfill. This includes, among other things:
The intended purpose therefore describes the areas (e.g. diagnostics or therapy) and the objectives (e.g. healing, support or prevention) for which the product is used.
The mechanism of action of a medical device describes the way in which the device works. The MDR specifically points out that the mechanism of action not based on pharmacological, immunological or metabolic effects. This means that the product does not work by taking medication or by influencing bodily functions such as the immune system or metabolism. Instead, it works via physical or chemical processes.
For example, a pacemaker uses physical effects to stimulate the heart, but it does not directly influence the metabolism or the immune system.
Accessories are products that are not medical devices themselves, but have been specially developed by the manufacturer to be used together with one or more medical devices. The accessory supports the use of the main product and helps to optimize the medical function of the product. It is therefore not a medical product in its own right, but rather a supplement to make the main product functional or to increase its efficiency.
The accessories can be used to ensure the intended purpose of the medical device, to adapt it for special applications, to facilitate handling, to increase performance or to combine the functions of the product with those of other devices.
For example, a tube that is integrated into a medical device such as a ventilator is an accessory. Without the tube, the device would not function as intended.
"Active devices" are medical devices whose operation requires an external energy source. This energy source cannot be human physical strength or gravity. These devices generate an active function that is necessary for the medical purpose of the device. Example:
"Systems and treatment units" consist of a combination of medical devices that are used together to fulfill a specific medical purpose. These products work together to support a complex medical function.
For example, a surgical system could consist of multiple instruments, such as a surgical robot, cameras and other supporting devices that work together to perform an operation.
Such systems and treatment units can comprise several products that are connected or integrated in a certain way to ensure successful treatment.
The "intended purpose" of a product refers to the use that the manufacturer has intended for the product. This is defined on the labeling of the product and in the instructions for use as well as in advertising or sales information. It describes what the product is intended to do and how it is used, for example:
The intended purpose therefore specifies the medical context in which the product is to be used and the purpose of its use.
Within the framework of the MDR, there are defined procedures to clarify how a product should be classified and treated. The status of a product is determined by the European Commission. It decides whether a product is a medical device and what status it has.
Status determination can be initiated in the following ways:
- At the request of a Member State: A Member State can request a review from the Commission if there is a good reason to do so (e.g. ambiguities about the classification of the device).
- On the Commission's own initiative:The Commission can also initiate a review on its own initiative if it considers that this is necessary (e.g. for new devices or technologies).
- After consulting the Medical Device Coordination Group: Before the Commission takes a final decision, it will consult a group of experts (Coordination Group), which will advise it.
These steps serve to guarantee legal certainty and ensure that products are correctly classified, which in turn guarantees the safety and protection of the health of EU citizens.